Updating the Genome Decoder
Later: Nucleotide Repetition Lengths
Earlier: Getting Our Hands Dirty (with the Human Genome)
In our last post we saw that so-called “N-blocks” (regions of the genome for which sequences are not available) were not getting decoded correctly by our decoder.
The solution is to look back into the so-called “file index” which specifies where the N-blocks are, and how long each block is. The modified decoder looks like this:
(defn genome-sequence
"
Read a specific sequence, or all sequences in a file concatenated
together; return it as a lazy seq.
"
([fname]
(let [sh (sequence-headers fname)]
(lazy-mapcat (partial genome-sequence fname) sh)))
([fname hdr]
(let [ofs (:dna-offset hdr)
dna-len (:dna-size hdr)
byte-len (rounding-up-divide dna-len 4)
starts-and-lengths (get-buffer-starts-and-lengths ofs 10000 byte-len)]
(->> (for [[offset length] starts-and-lengths
b (read-with-offset fname offset length)]
(byte-to-base-pairs b))
(apply concat)
(take dna-len)
(map-indexed (fn [i x] (if (is-in-an-n-block i hdr)
:N
x)))))))
The primary modification is the map-indexed bit at the end, which
looks up the position of the base pair in the index using the
is-in-an-n-block function:
(defn is-in-an-n-block
([x hdr]
(let [{:keys [n-block-starts n-block-sizes]} hdr]
(is-in-an-n-block x n-block-starts n-block-sizes)))
([x n-block-starts n-block-lengths]
(let [pairs (map (fn [a b] [a (+ a b)])
n-block-starts n-block-lengths)]
(some (fn [[a b]] (< (dec a) x b)) pairs))))I tested the new decoder against FASTA files of the first two chromosomes of the human genome using the same procedure we used for yeast in my validation post. The N-blocks (and all other sequences) were decoded correctly.
The astute reader will note the use of lazy-mapcat instead of =mapcat:
(defn lazy-mapcat
"
Fully lazy version of mapcat. See:
http://clojurian.blogspot.com/2012/11/beware-of-mapcat.html
"
[f coll]
(lazy-seq
(if (not-empty coll)
(concat
(f (first coll))
(lazy-mapcat f (rest coll))))))
A version of genome-sequence which used mapcat instead of lazy-mapcat
worked fine for individual chromosomes (file sections), but
consistently ran out of memory when processing whole genome files. It
took a bit of research and hair-pulling to figure out that there is a
rough edge with mapcat operating on large lazy sequences.
Lazy sequences are a strength of Clojure in general – they allow one
to process large, or even infinite, sequences of data without running
out of memory, by consuming and emitting values only as needed, rather
than all at once. Many of the core functions and macros in Clojure
operate on sequences lazily (producing lazy sequences as output),
including map, for, concat, and so on. mapcat is among these; however,
mapcat is apparently not “maximally lazy” when concatenating other
lazy seqs, causing excessive memory consumption as explained in this
blog post. Using that post’s fully lazy (if slightly slower) version
of mapcat fixed my memory leak as well. Though lazy seqs are awesome
in many ways, one does have to be careful of gotchas such as this one.
So, in the process of handling this relatively large dataset, we have
discovered two rough edges of Clojure, namely with mapcat and
count. We shall see if other surprises await us. Meanwhile, the latest
code is up on GitHub.
Back to Frequencies
Now that we are armed with a decoder which handles N-blocks correctly, let us return to our original problem: nucleotide frequencies. For yeast, we again have:
jenome.core> (->> yeast genome-sequence frequencies)
{:C 2320576, :A 3766349, :T 3753080, :G 2317100}Same as last time. For humans,
jenome.core> (->> human genome-sequence frequencies)
{:N 239850802, :T 856055361, :A 854963149, :C 592966724, :G 593325228}As expected, the GAC numbers are the same as what we had previously; only T and N have changed. The distributions look like so:
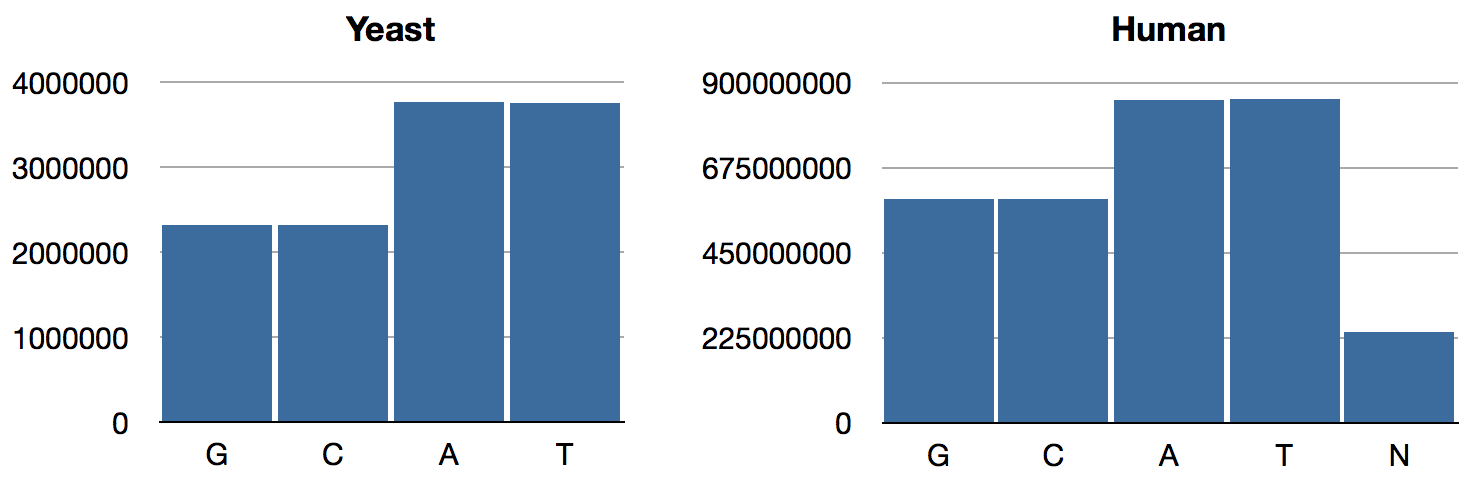
We can see that Chargaff’s Rule obtains now: A/T ratios are equal, as are G/C ratios. (BTW, this rule is intuitively obvious if you consider that in the double-helix structure, As are paired with Ts and Gs are paired with Cs.) Interestingly, the relative abundance of GC pairs in the human genome is higher than it is in yeast.
Timing and Parallelizing
Many of these “queries” on our data take a long time to run (particularly for the human genome). So as not to tie up my REPL and preventing me from doing other (presumably shorter) experiments, I find it helpful to run such tasks inside a little macro (similar to Clojure’s time) which performs the computation in a thread and, when the task is finished, prints the time elapsed, the code that was run, and the result:
(defmacro tib
"
tib: Time in the Background
Run body in background, printing body and showing result when it's done.
"
[expr]
`(future (let [code# '~expr
start# (. System (nanoTime))]
(println "Starting" code#)
(let [result# ~expr
end# (. System (nanoTime))
dursec# (/ (double (- end# start#)) (* 1000 1000 1000.0))]
(println (format "Code: %s\nTime: %.6f seconds\nResult: %s"
code#
dursec#
result#))
result#))))
For example, our overflowing count runs as follows:
(tib (count (range (* 1000 1000 1000 3))))
Starting (count (range (* 1000 1000 1000 3)))
;; ...
Code: (count (range (* 1000 1000 1000 3)))
Time: 399.204845 seconds
Result: -1294967296Another tool which has proved useful is:
(defmacro pseq
"
Apply threading of funcs in parallel through all sequences specified
in the index of fname.
"
[fname & funcs]
`(pmap #(->> (genome-sequence ~fname %)
~@funcs)
(sequence-headers ~fname)))
This threads one or more funcs through all the sequences in the file,
as mapped out by the file headers. The magic here is pmap, which is
map parallelized onto separate threads across all available
cores. pseq maxes out the CPU on my quad-core Macbook Pro and gives
results significantly faster:
Code: (->> yeast genome-sequence frequencies)
Time: 33.090855 seconds
Result: {:C 2320576, :A 3766349, :T 3753080, :G 2317100}
Code: (apply (partial merge-with +) (pseq yeast frequencies))
Time: 16.056695 seconds
Result: {:C 2320576, :A 3766349, :T 3753080, :G 2317100}With our updated decoder and these new tools, we can continue our poking and prodding of the genome in subsequent posts.
Later: Nucleotide Repetition Lengths
Earlier: Getting Our Hands Dirty (with the Human Genome)